前面我們有介紹了利用 10x Space Ranger 軟件分析空間轉(zhuǎn)錄組原始數(shù)據(jù)得到可用于下游分析的矩陣和鏡像文件。今天來介紹一下怎么利用 Space Ranger 的結(jié)果文件進(jìn)行后續(xù)分析,這里主要使用 Seurat 在進(jìn)行下游分析。
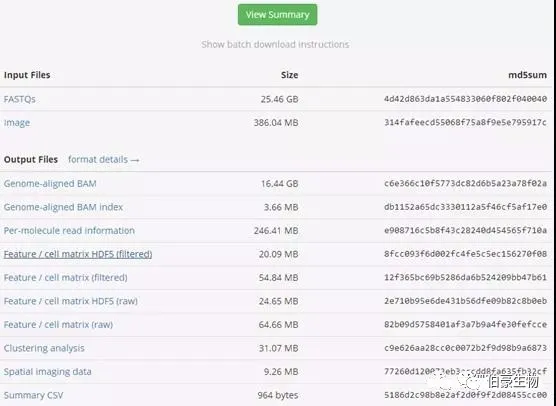
先來回顧一下跑完 Space Ranger 得到哪些結(jié)果文件:
Outputs:
|
用 seurat 進(jìn)行下游分析主要用到兩個(gè)結(jié)果文件。一個(gè)是 filtered_feature_bc_matrix.h5 文件,一個(gè)是 spatial 鏡像結(jié)果目錄。
安裝 R 包
由于 seurat 分析空間轉(zhuǎn)錄組的 R 包 satijalab-seurat 是在 GitHub 上的,如果我們需要直接安裝的話需要先安裝 R 包 devtools,然后利用 devtools 工具中的 install_github 來安裝 GitHub 上的 R 包。
安裝 devtools
install.packages('devtools')安裝 satijalab-seurat
devtools::install_github("satijalab/seurat",ref = "spatial")考慮到直接安裝 github 上的 R 包速度是很慢的,非常考驗(yàn)網(wǎng)速,可能需要多次才能安裝成功,我們也可以直接下載安裝包,本地安裝。
# 下載https://codeload.github.com/satijalab/seurat/legacy.tar.gz/spatial# 安裝install.packages("satijalab-seurat-v3.1.5-351-g85610bc.tar.gz", repos = NULL, type = "source")
注意該 R 包還在開發(fā)中,不要和之前安裝的 seurat 包沖突。
數(shù)據(jù)準(zhǔn)備
這里使用從 10x 官網(wǎng)下載的小鼠腦組織樣本 MouseBrain Serial Section 1 (Sagittal-Posterior)。
下載網(wǎng)址:https://support.10xgenomics.com/spatial-gene-expression/datasets
點(diǎn)擊選擇的樣本,下載兩個(gè)數(shù)據(jù)就行:
cell matrix HDF5 (filtered)和 Spatial imaging data
導(dǎo)入 R 包,讀取文件
library("Seurat")library("ggplot2")library("cowplot")library("dplyr")library("hdf5r")## 讀取矩陣文件name='Posterior1'expr <- "/pubj/ST_test/RNA/Sagittal-Posterior1/V1_Mouse_Brain_Sagittal_Posterior_filtered_feature_bc_matrix.h5"expr.mydata <- Seurat::Read10X_h5(filename = expr)mydata <- Seurat::CreateSeuratObject(counts = expr.mydata, project = 'Posterior1', assay = 'Spatial')mydata$slice <- 1mydata$region <- 'Posterior1' #命名# 讀取鏡像文件imgpath <- "/pubj/ST_test/RNA/Sagittal-Posterior1/spatial"img <- Seurat::Read10X_Image(image.dir = imgpath)Seurat::DefaultAssay(object = img) <- 'Spatial'img <- img[colnames(x = mydata)]mydata[['image']] <- imgmydata #查看數(shù)據(jù)An object of class Seurat32285 features across 3355 samples within 1 assayActive assay: Spatial (32285 features)
從 mydata 的輸出信息我們可以知道,這個(gè)樣本包含 3355 個(gè) spot 點(diǎn)、32285 個(gè)基因。
基礎(chǔ)統(tǒng)計(jì)作圖
##UMI 統(tǒng)計(jì)畫圖plot1 <- VlnPlot(mydata, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "nCount_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)
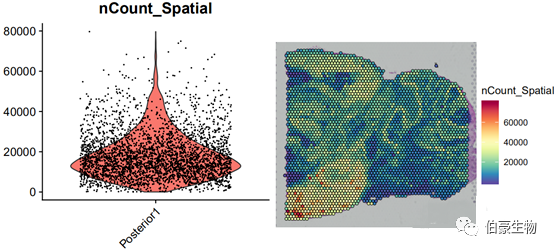
UMI 數(shù)大部分集中到 10000-20000 區(qū)間,不超過 80000,并且組織中高 UMI 數(shù)的區(qū)域主要集中在左下角。后面可以關(guān)注一下左下角區(qū)域的基因的表達(dá)和主要的細(xì)胞類型。
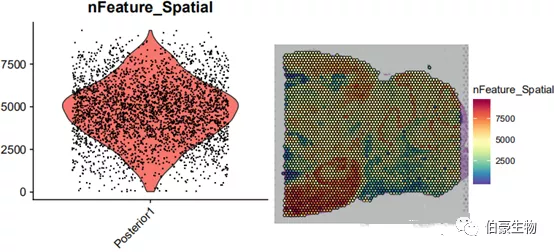
##gene 數(shù)目統(tǒng)計(jì)畫圖plot1 <- VlnPlot(mydata, features = "nFeature_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "nFeature_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)
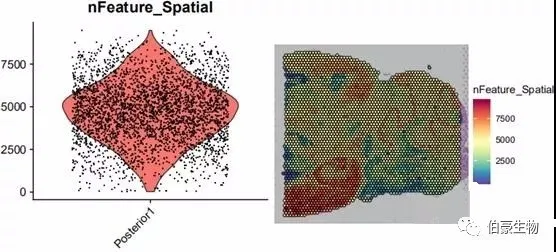
基因數(shù)目大部分處于 2500-7500 之間,結(jié)合 UMI 數(shù)據(jù)的分布可以發(fā)現(xiàn) UMI 數(shù)目高的區(qū)域基因數(shù)也高,說明基因數(shù)和 UMI 數(shù)基本上是呈正相關(guān)的。
# 線粒體統(tǒng)計(jì)mydata[["percent.mt"]] <- PercentageFeatureSet(mydata, pattern = "^mt[-]")plot1 <- VlnPlot(mydata, features = "percent.mt", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "percent.mt") + theme(legend.position = "right")plot_grid(plot1, plot2)
注意如果是人的數(shù)據(jù) pattern ="^mt[-] 改成 pattern ="^MT[-]
總體來說,這個(gè)樣本的線粒體比例不高,左邊中上區(qū)域有一處線粒體比例稍微高一點(diǎn),后面也可以仔細(xì)研究一下這一塊區(qū)域到底是特定的細(xì)胞類型引起的還是組織活性的差異引起的。不過從這張圖我們還可以發(fā)現(xiàn)一個(gè)有意思的現(xiàn)象,基因和 UMI 高表達(dá)的區(qū)域往往線粒體比例更低。
數(shù)據(jù)過濾
做單細(xì)胞 RNAseq 我們都會(huì)根據(jù) UMI、基因數(shù)、線粒體比例等進(jìn)行過濾,那么做空間轉(zhuǎn)錄組數(shù)據(jù)分析其實(shí)我們也可以按這樣的方式來過濾。具體的過濾條件需要根據(jù)具體樣本數(shù)據(jù)來定,沒有固定的標(biāo)準(zhǔn)。
比如這個(gè)樣本我們可以設(shè)置過濾條件:
①基因數(shù)大于 200,小于 7500
②UMI 數(shù)大于 1000,小于 60000
③線粒體比例小于 25%
mydata2 <- subset(mydata, subset = nFeature_Spatial> 200 & nFeature_Spatial <7500 & nCount_Spatial> 1000 & nCount_Spatial <60000 & percent.mt < 25)mydata2An object of class Seurat32285 features across 2977 samples within 1 assayActive assay: Spatial (32285 features)
過濾后還剩 2977 個(gè) spot 點(diǎn)。過濾后我們?cè)诶L制一下 UMI 分布圖。
plot1 <- VlnPlot(mydata2, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata2, features = "nCount_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)

那么現(xiàn)在問題來了,過濾之后組織圖像里面缺了幾塊,顯得特別丑。那么我們到底應(yīng)不應(yīng)該過濾呢?過濾數(shù)據(jù)可以減少離群的點(diǎn),減少對(duì)后面聚類結(jié)果的影響,不過濾數(shù)據(jù)可以讓組織圖像保持完整性,繪圖更好看一點(diǎn),所以這個(gè)還真不好決斷。
數(shù)據(jù)歸一化
Seurat 官方推薦使用 sctransform 歸一化方法,它構(gòu)建了基因表達(dá)的正則化負(fù)二項(xiàng)模型,以便在保留生物差異的同時(shí)考慮技術(shù)因素。
Sctransform 函數(shù)同時(shí)實(shí)現(xiàn)了 NormalizeData、ScaleData、FindVariableFeatures 三個(gè)函數(shù)的功能。
mydata <- SCTransform(mydata, assay = "Spatial", verbose = FALSE)基因表達(dá)可視化
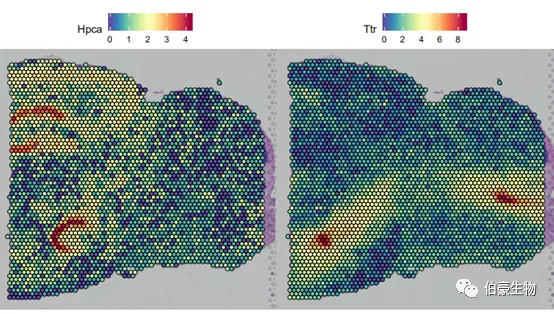
Seurat 的 SpatialFeaturePlot 功能擴(kuò)展了 FeaturePlot,可以將表達(dá)數(shù)據(jù)覆蓋在組織組織上。這里展示的 Hpca 基因是一個(gè)強(qiáng)的海馬 marker,Ttr 是一個(gè)脈絡(luò)叢 marker。可以通過基因的表達(dá)分布來初步判斷一下海馬區(qū)和脈絡(luò)叢區(qū)處于組織切片的哪個(gè)位置。
SpatialFeaturePlot(mydata, features = c("Hpca", "Ttr"))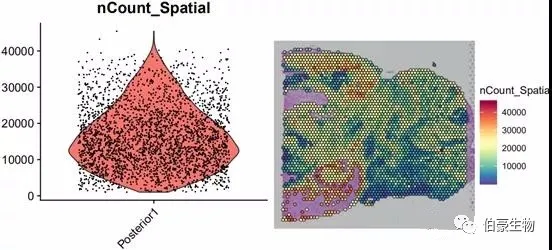
從結(jié)果的展示來看,這兩個(gè) marker 基因的分布還是挺集中的,這也說明理由空間轉(zhuǎn)錄組數(shù)據(jù)來分析小鼠腦的不同區(qū)域的表達(dá)差異應(yīng)該還是比較準(zhǔn)確的。另外,海馬區(qū)的分布可以大概分成 3 大塊,從上之下首塊弧形區(qū)域似乎處于線粒體高表達(dá)區(qū)域,而下面一塊弧形區(qū)處于基因高表達(dá)區(qū)。后面可以把這三個(gè)不同區(qū)域的數(shù)據(jù)進(jìn)行差異基因和功能的比較也許會(huì)發(fā)現(xiàn)一些有意思的東西。
降維、聚類和可視化
接下來利用 seurat 進(jìn)行降維和聚類。先進(jìn)行 PCA 降維,再選擇前 30 個(gè)維度進(jìn)行聚類和 umap、tsne 降維。
mydata <- RunPCA(mydata, assay = "SCT", verbose = FALSE)mydata <- FindNeighbors(mydata, reduction = "pca", dims = 1:30)mydata <- FindClusters(mydata, verbose = FALSE)mydata <- RunUMAP(mydata, reduction = "pca", dims = 1:30)mydata <- RunTSNE(mydata, reduction = "pca",dims = 1:30)
tsne 展示結(jié)果:
Umap 展示結(jié)果:
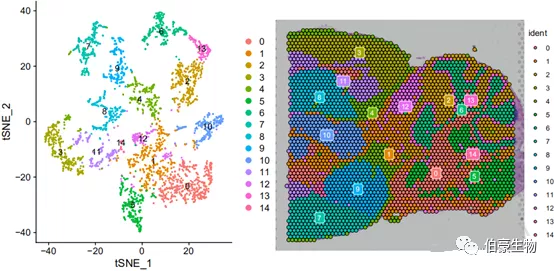
tsne 和 umap 兩種展示方式在這次分析里差別不是特別大,tsne 相對(duì)來說亞群與亞群之間分的更開,而 umap 則單個(gè)亞群位置更集中。這個(gè)時(shí)候我們也可以結(jié)合前面 marker 基因的表達(dá)分布圖來大概判斷一下每個(gè)亞群大概處于小鼠腦的那個(gè)區(qū)。
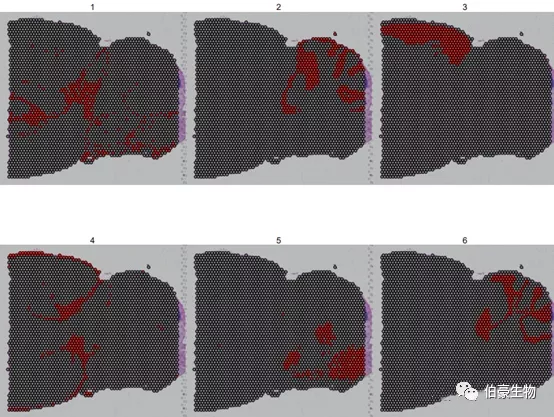
由于亞群的顏色比較接近,有時(shí)候不太好判斷,我們可以是 cells.highlight 來標(biāo)記特定的亞群。
SpatialDimPlot(mydata, cells.highlight = CellsByIdentities(object = mydata, idents = c(1, 2, 3, 4,5, 6)), facet.highlight = TRUE, ncol = 3)
今天先分享到這,下期繼續(xù)!
更多伯豪生物人工服務(wù):

